


Magally M. Núñez Naranjo.
RESUMEN
Las porfirias constituyen un grupo de trastornos causados por defectos en la vía de síntesis del grupo hemo. En la porfiria eritropoyética congénita existe deficiencia de la uroporfirinógeno sintetasa, lo que a su vez provoca acumulación de grandes cantidades de uroporfirina I en todos los tejidos; dando lugar a fotosensibilidad con lesiones mutilantes de la piel, eritrodoncia, anemia hemolítica, esplenomegalia, y fragilidad ósea. El diagnóstico definitivo se fundamenta en la demostración de la actividad deficiente de la uroporfirinógeno sintetasa, o la determinación de mutaciones específicas en el gen respectivo. El rasgo histopatológico característico muestra una ampolla subepidérmica y su tratamiento requiere colaboración multidisciplinaria. En el presente artículo se describe un caso de porfiria eritropoyética congénita con la presentación clínica dermatológica clásica.
Palabras clave: porfirinas; síntesis grupo hemo; porfiria; uroporfirinógeno -III sintetasa
ABSTRACT
Congenital erythropoietic porphyria. Case report
Porphyrias are a group of disorders caused by defects in the synthesis pathway of heme.
Congenital erythropoietic porphyria is characterized by uroporphyrinogen synthase deficiency, which causes accumulation of large amounts of uroporphyrin I in all tissues; resulting in photosensitivity with mutilating skin lesions, erythrodontia, hemolytic anemia, splenomegaly, and bone fragility. Definitive diagnosis is based on demonstrating poor uroporphyrinogen synthase activity, or determination of specific mutations in the respective gene. Characteristic histopathological feature shows a subepidermal blister and treatment requires multidisciplinary collaboration.
A case of congenital erythropoietic porphyria with classical dermatological clinical presentation is reported.
Key words: porphyrins; heme biosynthesis; porphyria; uroporphyrinogen-III synthase
► INTRODUCCIÓN
Las porfirias son un grupo de enfermedades metabólicas causadas por deficiencias en las enzimas de la vía de síntesis del grupo hemo. En función de la enzima alterada, se acumulan los distintos sustratos (precursores y porfirinas) que desencadenan las manifestaciones clínicas propias de cada porfiria. Se han descrito mutaciones en 7 genes: aminolevulinato deshidratasa (ALAD), porfobilinógeno desaminasa (PBGD), uroporfirinógeno sintetasa (UROS), uroporfirinógeno decarboxilasa (UROD), coproporfirinóge-
no oxidasa (CPOX), protoporfirinógeno oxidasa (PPOX) y ferroquelatasa (FECH), responsables de los 7 tipos de porfiria reconocidos. Sin embargo, en los últimos años se ha demostrado la participación de otros genes que codifican para proteínas, que no forzosamente forman parte de la vía de biosíntesis del grupo hemo como causantes de formas concretas de porfiria1.
La porfiria eritropoyética congénita (PEC), o enfermedad de Günther1, es una enfermedad autosómica recesiva rara del metabolismo de la porfirina2, en la cual el defecto genético es la deficiencia pero no ausencia3 de la enzima UROSIII, codificada por el gen UROS localizado en el cromosoma 10q25.3→q26.31. La mutación C73R causa la más severa deficiencia de la enzima UROS4.
Esta enfermedad se manifiesta generalmente en la lactancia o la primera infancia, aunque puede ser diagnosticada más tadíamente2. Su clínica, aunque heterogénea5, responde a la acumulación de uroporfirina I y coproporfirina I3 en la piel, eritrocitos, médula ósea, dientes, huesos y otros órganos6. Se caracteriza por fotosensibilidad cutánea grave con formación de ampollas6 que llevan a la mutilación de los tejidos con pérdida de los rasgos faciales; además se acompaña de hipertricosis y alopecia3. Los depósitos de porfirinas en la córnea y en los dientes en formación pueden producir defectos visuales y eritrodoncia respectivamente. La presencia de anemia hemo- lítica es muy frecuente, sólo en raras ocasiones puede estar ausente6. Los pacientes suelen mostrar anisocitosis, poiquilocitosis, reticulocitosis, ausencia de haptoglobina, hiperbilirrubinemia y aumento del urobilinógeno fecal. El hiperesplenismo secundario, que se produce por eritropoyesis ineficaz, contribuye a la anemia y puede producir leucopenia y trombocitopenia adicionalmente1.
También se distingue porfirinuria masiva, evidenciada por orina de color rojo6, anormalidades esqueléticas como acroosteólisis con mutilaciones, osteodistrofia y osteoporosis por aumento de recambio5.
► CASO CLÍNICO
Paciente de 7 años de edad, único hijo de padres no consanguíneos, con antecedentes personales de anemia desde el nacimiento y madre con diabetes mellitus tipo 2.
Consulta en el servicio de Dermatología por presentar desde su nacimiento ampollas recurrentes en áreas de piel fotoexpuestas, que llevan a la formación de cicatrices y eventual pérdida de uñas; asociada a la eliminación de orina color rojo y anemia persistente.
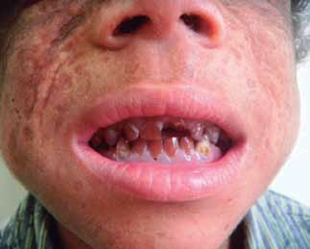
Al examen físico presenta dermatosis localizada en áreas de piel fotoexpuesta, caracterizada por la presencia de vesículas, placas costradas, cicatrices atróficas defor- mantes; en cara y extremidades se observa hipertricosis (Fig. 1). Además deformidad y limitación del movimiento en articulaciones interfalángicas de manos (Fig. 2), eritro- doncia (Fig. 3) y orina color rojo oscuro (Fig. 4). La explora- ción abdominal y neurológica fue normal.
Las investigaciones de laboratorio incluyeron pruebas de función tiroidea, hepática y renal, las cuales se encontraron dentro de límites normales, excepto Hb 11.4 g / dl,MCV 74.9 fL, MCH 23.3 pg. El examen elemental de orina indica bilirrubinas (++/+++) y urobilinógeno 12mg/dl. La radiografía de manos y pies muestra signos de retraso en el crecimiento óseo en relación a la edad cronológica del paciente e hipoplasia falángica distal bilateral; la de tórax exhibe cardiomegalia GII (expensas de cavidades izquierdas). La ecografía abdominal fue normal.
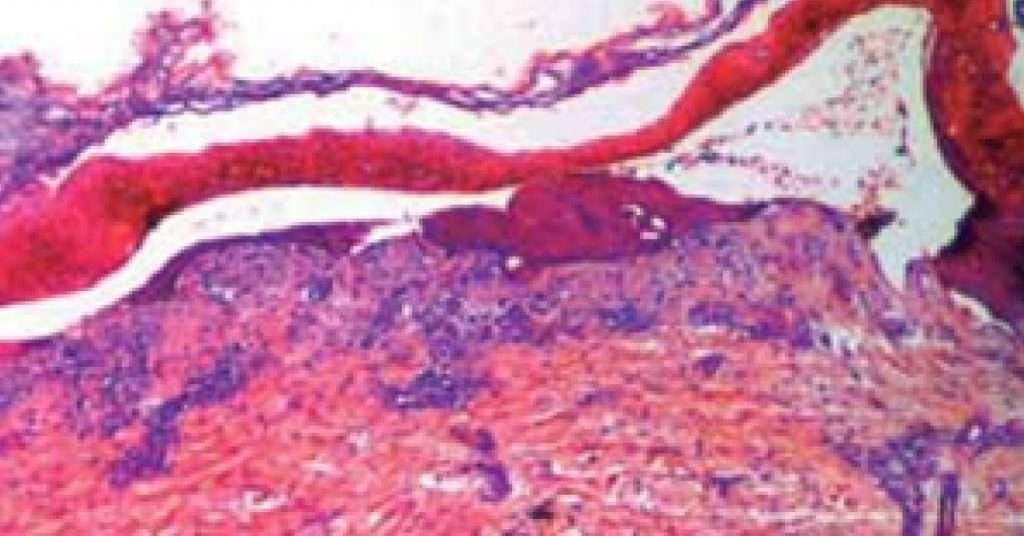
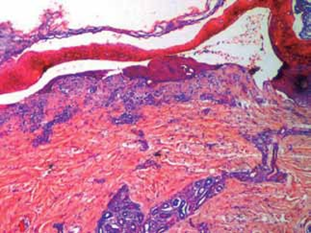
Se realiza biopsia de piel, la cual exhibe vesícula subepidérmica con escaso infiltrado linfocitario perivascular superficial (Fig. 5).
Sobre la base de los hallazgos clínicos y de laboratorio realizamos el diagnóstico de porfiria eritropoyética congénita. El paciente fue manejado interdisciplinariamente, con estricta evitación de la luz solar, prescripción de fotoprotector solar tópico y beta- caroteno oral. Complementariamente se indicó evaluación oftalmológica, la cual no señala patología asociada; pediatría determinó desnutrición GII y anemia microcítica hipocrómica. Finalmente fue referido al servicio de Genética, Hematología y Odontología en un hospital de mayor complejidad, con la finalidad de realizar demás estudios de laboratorio, ya que en el nuestro no fueron posibles por falta de infraestructura.
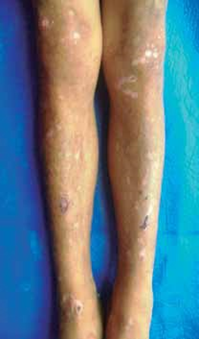
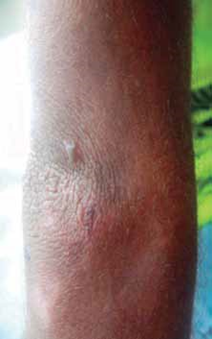
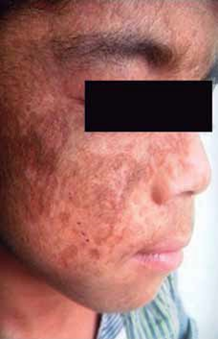

Fig. 2: acrogrifosis en articulaciones interfalángicas de manos. Fig. 3: eritrodoncia. Fig. 5:(HE 4x) vesícula subepidérmica.

► DISCUSIÓN
La porfiria eritropoyética congénita (PEC) tiene una fre- cuencia estimada de 1 en cada 2 a 3 millones de personas. A partir de 1997 se habían registrado unos 130-150 casos1. Debe sospecharse de PEC ante un recién nacido que presente orina de color rojo oscuro que mancha el pañal3 y posteriormente muestre eritrodoncia, siendo esta última prácticamente patognomónica de PEC6.
La presencia de niveles altos de uroporfirinas I en orina y eritrocitos son diagnósticos de PEC5. El diagnóstico se confirma mediante la demostración de la actividad marcadamente deficiente de la UROS o la identificación de mutaciones específicas en el gen UROS, situación que en nuestro paciente no fue posible debido a la falta de instalaciones de laboratorio. La enfermedad también se puede detectar intraútero mediante la medición de porfirinas en el líquido amniótico y la actividad UROS en las células amnióticas o vellosidades coriónicas cultivadas, o por la detección de mutaciones de la UROS específica de la familia3.
El diagnóstico diferencial de esta entidad debe realizarse con otras fotodermatosis congénitas de la infancia, como el xeroderma pigmentoso, hidroa vacciniforme, epidermólisis ampollosas hereditarias y con el penfigoide ampolloso infantil5.
Histopatológicamente el rasgo característico es una ampolla subepidérmica con un leve infiltrado linfocitario perivascular superficial9.
El manejo terapéutico se basa en tres pilares básicos: evitar la fotoexposición, el cuidado meticuloso de las heridas cutáneas y transfusiones sanguíneas y otras medidas de soporte hematológico1.
La evitación absoluta de la exposición al sol es crucial10, por lo tanto se recomienda el uso de protección solar tópica6, ropa protectora, gafas solares, películas protectoras en las ventanas, bombillas incandescentes de color rojizo y pantallas de filtrado para luces fluorescentes11.
Las infecciones bacterianas cutáneas requieren un tratamiento oportuno para evitar cicatrices y mutilaciones. También es importante proteger la piel de probables traumatismos5.
La lubricación tópica de los ojos mejora los síntomas de ojo seco y puede estabilizar la función visual 10.
Las medidas orales que se han utilizado incluyen betacaroteno, carbón activado y colestiramina para interrumpir y prevenir la reabsorción de porfirinas, pero las grandes dosis requeridas de estos agentes a menudo hacen su uso poco práctico.
Se recomienda el uso de alfatocoferol oral y ácido ascórbico para reducir el efecto de los radicales de oxígeno reactivo sobre elementos de la piel y eritrocitos circulantes sensibilizados11.
La esplenectomía disminuye la anemia hemolítica por el aumento de la esperanza de vida de los eritrocitos; sin embargo, los beneficios son de corta duración. En los casos más graves (pacientes transfusión-dependientes) el trasplante hematopoyético es el tratamiento de elección1, pero los resultados a largo plazo son desconocidos y las complicaciones infecciosas potencialmente mortales limitan la aplicabilidad de este enfoque terapéutico.
La depilación láser se puede utilizar para tratar la hipertricosis facial, mientras que la intervención quirúrgica está indicada en caso de mutilación grave (reparación de microstomía, corrección del ectropión, reconstrucción de la nariz)13.
Pese a que aún no se dispone de una terapia génica para la curación de esta patología, existen logros relevan- tes con el uso de lentivectores terapéuticos que parecen augurar una herramienta esperanzadora e interesante en el manejo de los pacientes afectados16.
► CONCLUSIÓN
El objetivo de este artículo es compartir nuestra experien- cia con un tipo raro de porfiria cutánea que exhibe una
sintomatología dermatológica florida que, para muchos, no ha podido ser observada durante toda su vida profesional. En este caso en particular no se pudo contar con la demostración de la actividad deficiente de la uroporfirinógeno sintetasa, ni la ubicación de mutaciones específicas en el gen comprometido que, según la literatura, son consideradas valiosas en el diagnóstico. Además poner una vez más en evidencia que la adecuada anamnesis y exploración del paciente es esencial en el ejercicio médico habitual.
► BIBLIOGRAFÍA
- Darwich, E.; Herrero, C.: Novedades en las porfirias eritropoyé- ticas. Actas Dermosifiliogr 2013; 104: 212-219.
- Baran, M.; Eliaçık, K.; Kurt, I.; Kanık, A.; Zengin, N.; Bakiler, A.R.: Bullous skin lesions in a jaundiced infant after photothe- rapy: a case of erythropoietic porphyria. Turk J Pediatr. 2013; 55: 218-221.
- Balwani, M.; Desnick, R.J.: The porphyrias: advances in diag- nosis and treatment. Hematology Am Soc Hematol Educ Pro- gram. 2012; 2012:19-27.
- Berry, A.A.; Desnick, R.J.; Astrin, K.H.; Shabbeer, J.; Lucky, A.W.; Lim, H.W.: Two brothers with mild congenital erythropoie- tic porphyria due to a novel genotype. Arch Dermatol 2005; 141:1575-1579.
- Muñoz, C.; Herrero Mateu, C.: Porfirias cutáneas. Med Cutan Iber Lat Am 2005; 33:193-210.
- De, A.K.; Das, K.; Sil, A.; Joardar, S.A.: Case of Congenital Erythropoietic Porphyria without Hemolysis. Indian J Dermatol 2013; 58:407.
- Melito, V.A.; Rossetti, M.V.; Parera, V.E.; Batlle, A.: Porfirias poco frecuentes. Casos detectados en la población argentina. Rev Argent Dermatol 2006; 87: 248-263.
- Gucev, Z.; Slavevska, N.; Tasic, V.; Laban, N.; Pop-Jordano- va, N.; Danilovski, D.; Woolf, J.; Cole, D.: Congenital erythro- poietic porphyria with two mutations of the uroporphyrinogen III synthase gene (Cys73Arg, Thr228Met). Indian J Hum Ge- net 2011; 17: 104-107.
- Egbert, B.M.; LeBoit, P.E.; McCalmont, T.; Hu, C.H.; Austin, C.: Caterpillar bodies: distinctive, basement membrane-containing structures in blisters of porphyria. Am J Dermatopathol 1993; 15: 199-202.
- Mir, M.A.: Porphyria overview. emedicineSpecialities>Dermat ology> Diseases.emedicine from webmed; by Medscape [up- dated 2014 Mar 18; cited 2014 Nov 26]. Disponible en: http:// emedicine.medscape.com/article/1389981-overview#a11.
- Katugampola, R.P.; Anstey, A.V.; Finlay, A.Y.; Whatley, S.; Woolf, J.; Mason, N.; Deybach, J.C.; Puy, H.; Ged. C.; de Verneuil, H.; Hanneken, S.; Minder, E.; Schneider-Yin, X.; Badminton, M.N.: A management algorithm for congenital erythropoietic por- phyria derived from a study of 29 cases. Br J Dermatol 2012; 167: 888-900.
- Desnick, R.J.; Astrin, K.H.: Congenital erythropoietic porphyria: advances in pathogenesis and treatment. Br J Haematol 2002; 117: 779-795.
- Kauffman, L.; Evans, D.I.; Stevens, R.F.; Weinkove, C.: Bone- marrow transplantation for congenital erythropoietic porphyria. Lancet 1991; 337: 1510-1511.
- Tezcan, I.; Xu, W.; Gurgey, A.; Tuncer, M.; Cetin, M.; Oner, C.; Yetgin, S.; Ersoy, F.; Aizencang, G.; Astrin, K.H.; Desnick R.J.: Congenital erythropoietic porphyria successfully treated by allogeneic bone marrow transplantation. Blood 1998; 92: 4053- 4058.
- Harada, F.A.; Shwayder, T.A.; Desnick, R.J.; Lim, H.W.: Treatment of severe congenital erythropoietic porphyria by bone marrow transplantation. J Am Acad Dermatol 2001; 45: 279-282.
- Robert, E.; Lalanne, M.; Lamrissi, I.; Guyonnet, V.; Richard, E.; Pitard, V.; Mazurier, F.; Moreau-Gaudry, F.; Ged, C.; de Verneuil, H.: Modeling of congenital erythropoietic porphyria by RNA in- terference: a new tool for preclinical gene therapy evaluation. J Gene Med 2010; 12: 637-646.
